
Download Here: Troubleshooting GT_Recon Strategy 2022
In the last decade, gene therapies have been a major area of development and interest. What kicked off with the approval of Spark Therapeutics’ Luxturna in 2017 has now blossomed into a robust pipeline including the approval of Zolgensma (Novartis), and a number of cutting-edge therapies in clinical trials (such as Uniqure and CSL Behring’s therapy for hemophilia or Ultragenyx’s therapy for MPS IIIA). However, while the number of gene therapies entering clinical trials has increased, so have the inevitable stumbling blocks in development. Consequently, the FDA has brought greater attention to gene therapy safety, with a spate of clinical holds and culminating in a special Advisory Committee broadly covering AAV-based products. While no direct recommendations came from the committee, it highlighted the FDA’s specific concerns on the potential safety risks that are not yet well understood. At the same time, companies have responded in non-traditional ways to troubleshoot their drugs; particularly the recent collaboration of Pfizer, Sarepta, Genethon, and Solid Biosciences on Duchenne Muscular Dystrophy (DMD) gene therapy safety. In response to these rapidly occurring events, we took a deep dive into the current state of gene therapy, with a focus on consolidating the safety signals to date and the potential innovations that could mitigate these issues and accelerate future development.
An unprecedented industry collaboration
The partnership between Pfizer, Sarepta, Genethon, and Solid Biosciences is truly a first in the gene therapy field, and unique for the industry in general. All four companies are developing AAV gene therapies for DMD, and almost all saw serious adverse events (SAE) of muscle weakness in their trials. Rather than individually trying to determine the cause and leaving their rivals in the dark, the companies chose to work together and share relevant data. The competitive risk of sharing data seems to have paid off, and the working group found a potential explanation related to the mutational type of patients. In every patient with the therapy related muscle weakness, researchers found that their deletion contained a section of dystrophin seen on the transgene. Because the new protein had a portion never before seen in the body, it triggered a T cell response against the muscle cells in some of these patients. While this breakthrough provides an answer to the collaborating companies, it also creates a massive question for all gene therapy developers – how do they ensure their patients don’t react in a similar way to any new, functional protein?
Gene therapy effects expand outside the active plasmid
Outside of the transgene (literally), gene therapy vectors have seen scrutiny. Of the concerns outlined by the September FDA advisory committee, three of them (hepatotoxicity, thrombotic microangiopathy (TMA), and oncogenicity) have the potential to be impacted by vector choice. To better understand what hurdles gene therapy has to cross, we looked into these adverse effects and the existing potential solutions.
Hepatotoxicity is strikingly common in gene therapy patients: in Zolgensma trials 90/100 patients had elevated ALT/AST and roughly one-third had at least one hepatotoxicity adverse event. Perhaps due to this frequency, there is already a hypothesized mechanism – a T cell response to AAV capsids. In systemically delivered gene therapy, large numbers of viral vectors enter the liver and infect cells there, even if it’s not the intended target. This high level of infection triggers the immune response and leads to damage. This can be particularly concerning in conditions like spinal muscular atrophy (SMA) where patients already have liver damage, so ensuring the treatment can be tolerated is especially important.
TMA is less common than hepatotoxicity, and the etiology of the condition is less understood and defined. TMA is generally related to dysregulated complement activation, but the search for an exact cause is still frustrating researchers. Drugs and infections (including adenovirus (Ad)) are both known to lead to TMA, and while the gene therapies seen to cause TMA use AAV instead of Ad, the mechanism could be similar. There is also the chance that TMA in gene therapy can be familial due to complement mutations, and not only environmental. In any case, a response to the introduction of a large amount of virus necessary for gene therapy is likely to contribute to TMA.
Finally, there is also the potential for AAV-mediated oncogenicity in gene therapy patients. Regulators have already raised concerns over gene editing cancer risks, but AAV oncogenicity is separate. AAV therapies to date are engineered to avoid genome integration, but some can still randomly occur. Both humans and animal models have been shown to have viral DNA randomly distributed, but an oncogenic effect has only been seen in the livers of rodent models who already had damage from a high fat diet. While in vivo gene therapies have not yet caused cancer in a patient, the potential is still a fear nagging at the back of the mind for the FDA, even if it does not seem to be hindering approvals (see for example the recent vote on Bluebird Bio’s Eli-cel despite link to three cancer cases).
Necessity is the mother of innovation
For gene therapy vectors, they must, almost literally, carry the burden of getting the transgene where it needs to be, checking every box on a very long list. While the toxicities described above show there is more to be done, many advances have already been made to start addressing them – such as lowering the effective dose needed or finding alternative vector options.
No manufacturing process is 100% efficient, and gene therapy is no different. Impurities can be created at any step, including when packaging the plasmid. Many vectors do not uptake anything or just take up impurities; it’s been seen that 50-95% of AAV vectors from some manufacturing systems lack the desired plasmid[1]. While an empty capsid is not in itself toxic, they lower the concentration of effective vectors, and raise the chances of an immune response. Despite this issue, there are currently no official rules on capsid fill rates. The only guidance available is a draft submitted to the FDA this past May from Dark Horse, a gene therapy consulting group. Their recommended limit is based on what is currently used for cell therapy, that >70% of cells be alive, which correlates to less than 30% of capsids being empty in any gene therapy administration.
Encouragingly, it does seem like the industry is already paying attention to this issue. To address a clinical hold from the FDA, Solid Biosciences took multiple steps to improve their gene therapy for Duchenne’s (outside of collaborating as described above). One of these steps included improving their processing so that <10% of capsids were empty as compared to the original ~50%. Even more promising, based on findings published by Sartorius, up to 100% separation of the empty capsids can be reached[2]. Interestingly, both Solid Biosciences and Sartorius used types of chromatography to efficiently remove the empty capsids. Chromatographic separation is both efficient and scalable, which is essential to manufacture gene therapy, and there is a large variety of types (size exclusion, anion/canion, dual ion, etc.). Ultracentrifugation could also be used to remove product impurities, but it lacks scalability for the long-term due to the numerous steps and time commitment (30+ hours for two rounds)[3]. Going forward, it seems likely that chromatography will be the leading choice, and as tech advances we could expect empty capsid purification to meaningfully exceed Dark Horse’s suggested guideline.
An alternative approach to avoid viral vector toxicity is the potential use of non-viral vectors. Within the umbrella of non-viral vectors there is a full menu to choose from, with both physical and chemical options. Physical options include electroporation or sonoporation (see Table 1 for a longer list) and are in general considered to be potentially safer for patients, but with distinct limits in the location of administration and the efficiency of uptake. Another option is chemical vectors. Chemical administration of genetic material should be very familiar at this point – both the Pfizer and Moderna COVID-19 vaccines are made up of mRNA surrounded by a lipid protective layer. For gene therapy, lipid vectors are also an option, as well as polymers, dendrimers, polypeptides, and inorganic compounds. Each option has its own toxicity and efficiency (see Table 1 for a quick summary and Sharma et al. (2021) for more detailed descriptions), but as a general trend, as the efficiency of gene delivery increases, so does the toxicity of the compound.
Table 1: Example physical and chemical nonviral vector options
Adapted from Sharma et al. (2021)
| Vector type | Example Pros | Example Cons |
| Physical non-viral vectors | ||
| Electroporation | Proven safe and highly efficient administration | Only works in areas near electrodes
Electric pulses may damage DNA |
| Gene gun | Simple, fast, and highly efficient administration
No toxic chemical adjuvants Can deliver multiple genes simultaneously |
Only works in surface level tissues and lack of cellular target
Optimization required per cell type |
| Hydrodynamic injection | High efficiency, simplicity, safety, and reproducibility | Transgene level not controllable
Restricted clinical use due to large volume injection |
| Sonoporation | Known high safety, simplicity, and flexibility | Less effective than hydrodynamic injection and electroporation |
| Microneedle array | Works for skin conditions & is noninvasive yet efficient
Past clinical experience outlining efficient/safe use |
Hard to accurately measure dose
Incomplete migration of nucleic acid Cannot incorporate high loading dose |
| Magnetofection | Low chance of aggregation
Can transfect primary cells |
Low in vivo efficiency
Re-dosing leads to accumulation of iron oxide |
| Chemical vectors | ||
| Cationic lipids
(ex: DTOMA) |
Easily modulated for cell specificity
Relatively simple transfection procedure Enable endosomal escape |
Currently transfection efficiency too low to be clinically relevant |
| Cationic polymers
(ex: PEI) |
Water soluble
Can condense DNA more efficiently than lipids |
High cytotoxicity & non-biodegradable
Specific subtypes differ in exact cytotoxicity & degradability |
| Dendrimer based vectors
(ex: PPI) |
High gene transfection efficiency and low cytotoxicity
Protects from enzyme degradation |
Alterations required to improve cell targeting & compatibility |
| Polypeptide based vectors
(ex: MPG) |
Efficient targeting & cellular uptake | Inefficient endosomal release |
| Inorganic, polymeric, and lipid NP
(ex: quantum dots, SLN) |
Easily fine-tuned for desirable traits | Tradeoffs required in process of fine tuning (increased efficiency, increased toxicity, etc.) |
| Gemini surfactants | Alterable transfection efficiency and toxicity via head group, spacer, and tail modifications | Relatively newer and therefore may not be as well investigated |
Within nonviral vectors, electroporation has been a favorite for clinical use, but is mostly limited to ex vivo editing applications[4]. For in vivo gene therapy applications, polymer and lipid vectors have drawn attention so far. Examples include Celsion’s recent acquisition of EGEN, Inc. including their Theraplas system for modified PEI mediated vectors and Genprex’s positively charged lipid nanoparticles to deliver their oncological TUSC2 gene therapy. In the more preclinical, academic space, Johns Hopkins University has received attention for their biodegradable “nanocontainer” made up of 4 molecules and Imperial College London tried to use a liposome mediated vector for cystic fibrosis gene therapy (but more recently entered a deal with Boehringer Ingelheim for a lentiviral based version).
Charting the path forward
There are two major paths forward for vector development, but it remains to be seen if either becomes a clear preferred approach. Viral vectors have had thousands of years of evolution to perfect themselves compared to their non-viral counterparts, but in those thousands of years we have successfully fought off the corresponding infections and developed a strong immune response. Our natural reactions to viral infection are slowing down development, increasing costs, and making re-administration unlikely. While UK NICE has stated 40 years of durability is “uncertain but reasonable” for Zolgensma, it is not guaranteed and is still falling short of a full lifespan. Multiple deliveries of gene therapy could help extend therapeutic effect, but that is only possible if immunity can be avoided. Nonviral vectors can help to avoid immunity, but they struggle with transduction efficiency. Furthermore, developing nonviral vectors is expensive and may not be attractive in the near term, but this has to be balanced with the known high costs of viral vector commercial manufacturing. Overall, there is no simple answer to what vector to use or how to avoid immune responses (whether it be to the vector or the plasmid), but it is something that will be at the top of mind as the industry moves forward.
Figure 1: Illustrative progression of gene therapy
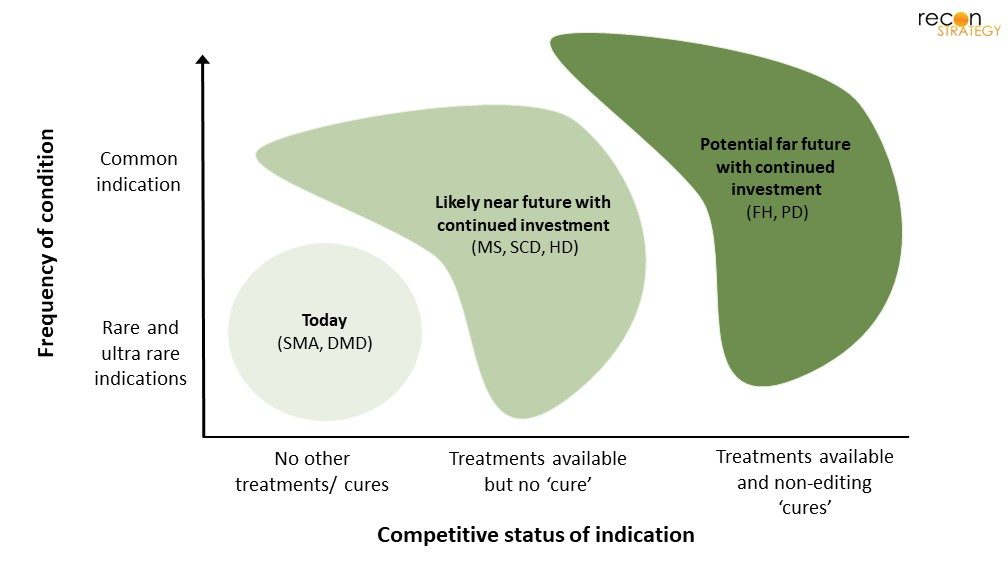
Similarly, there is no immediately available solution on how to address the transgene-related immune response. Increased profiling of exact patient mutations could be helpful, but there is also the risk that this is not 100% predictive and may limit patient pools. Not every patient with a deletion seen on the transgene will have an immune response and knocking these patients out of the treatment pool means that they will need to find alternative treatments (which are often not available). Lowering overall immunity may help, but it also will not be a panacea.
Overall, the high price tag of gene therapy development means that currently only very severe monogenic diseases, most of which are rare, are reachable by gene therapy; and the current risks, such as hepatotoxicity and TMA, mean that only those patients with no other effective options are well suited. If no progress is made to circumvent the aforementioned issues, it’s likely that gene therapy will be limited to these patients with the highest unmet need. However, if the current issues of gene therapy are mitigated, then more common and less severe diseases may be reachable (Figure 1)[5]. To reach this point though, continued investment is needed by companies to push forward vector development, both viral or non-viral. There are many examples of this continued innovation, from biotech companies that are building technologies for specific therapies (e.g., Generation Bio, Affinia Therapeutics) or broad-scale players that look to offer a best-in-class platform broadly to industry (e.g., Resilience). Assuming this investment in gene delivery technologies continues in the years ahead, we have high hopes for this modality, ultimately providing substantial benefit to patients and leading to a golden era of gene-based therapeutics.
Download Here: Troubleshooting GT_Recon Strategy 2022
[1] Wright, J. “Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment.” Biomedicines, vol. 2, no. 1, Mar. 2014, pp. 80–97. Crossref, https://doi.org/10.3390/biomedicines2010080.
[2] Gagnon, Pete, et al. “Removal of empty capsids from adeno-associated virus preparations by multimodal metal affinity chromatography.” Journal of Chromatography A 1649 (2021): 462210.
[3] Qu, Weihong, et al. “Scalable downstream strategies for purification of recombinant adeno-associated virus vectors in light of the properties.” Current pharmaceutical biotechnology 16.8 (2015): 684-695.
[4] Shim, Gayong et al. “Therapeutic gene editing: delivery and regulatory perspectives.” Acta pharmacologica Sinica vol. 38,6 (2017): 738-753. doi:10.1038/aps.2017.2
[5] Disease abbreviations: Spinal Muscular Atrophy (SMA), Duchenne’s Muscular Dystrophy (DMD), Multiple Sclerosis (MS), Sickle Cell Disease (SCD), Huntington’s Disease (HD), Familial Hypercholesterolemia (FH), Parkinson’s Disease (PD)