
Download a PDF of this article here.
Ex vivo gene-therapies have been a game-changer for aggressive hematologic cancers. These products can effectively cure patients who otherwise had very low chance of survival beyond 5 -years. However, making these products is exceptionally complex and the drugs are priced accordingly (from $370,000 to over $2.5M). While few patients receive these treatments today, clinical development could lead to a much broader population. If 1 in 20 newly diagnosed cancers were optimally treated with these high-price therapies, the cost to health-payers could become untenable (~$40B/yr in the US alone.)
We examine the state of development for these advanced therapeutics – exploring both autologous and allogeneic approaches. These modalities face distinct clinical and production challenges, and we highlight a few of the more interesting tactics being tested as solutions. Finally, we highlight why certain therapeutic areas are likely to shift to allogeneic, even if clinical risk remains, while others be justified in using autologous only
1. Why all the talk?
Ex vivo gene-therapies[1] (modified cell therapies such as Kymriah, Yescarta and Zynteglo) have become some of the most watched products in recent years. These drugs have had a remarkable impact on clinical care (e.g., Kymriah offers 5-year overall survival of 55% for relapsed or refractory B-cell ALL, compared to 10% with alternative care[2]), so it’s little surprise to see growing investment in the field. However, manufacture of these products is highly complex – all rely on patients’ own cells as key starting inputs (autologous) which leads to extreme prices ($370-420k for cancers, >$2M+ for rare-diseases[3]). If autologous cell therapies became commonly used, the budget impact would be massive. Consider, if these treatments became a first line treatment for hematologic cancers, the annual budget impact would exceed $35B in the US.[4]
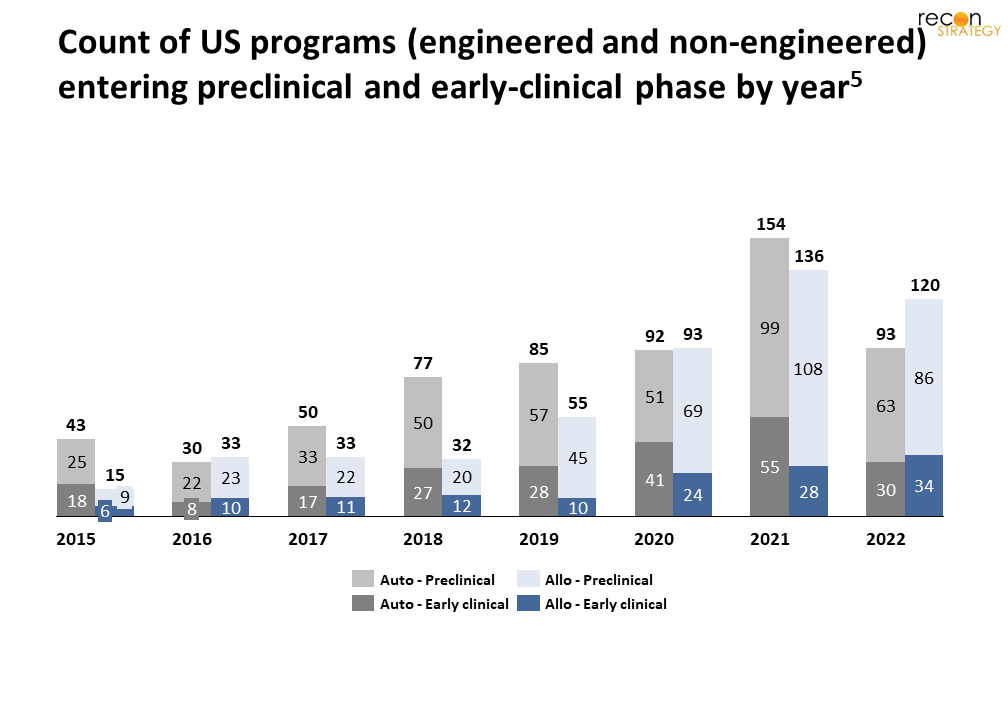
Investment in donor-cell derived (allogeneic) approaches has grown recently (Figure 1);[5] this kind of cell therapy might circumvent several production hurdles and lower production costs. However, many uncertainties remain about the safety and efficacy of allogeneic cells. Herein, we examine the state of development of both auto- and allo- cell therapies, outline clinical and production challenges, and highlight current approaches to address these issues. Our goal is not to provide a technical deep-dive, but rather offer a perspective on how these approaches can have a meaningful impact on accessibility, costs, and clinical outcomes, and if they favor auto- or allo- in the long-term.
2. A systematic framework to categorize cell therapies
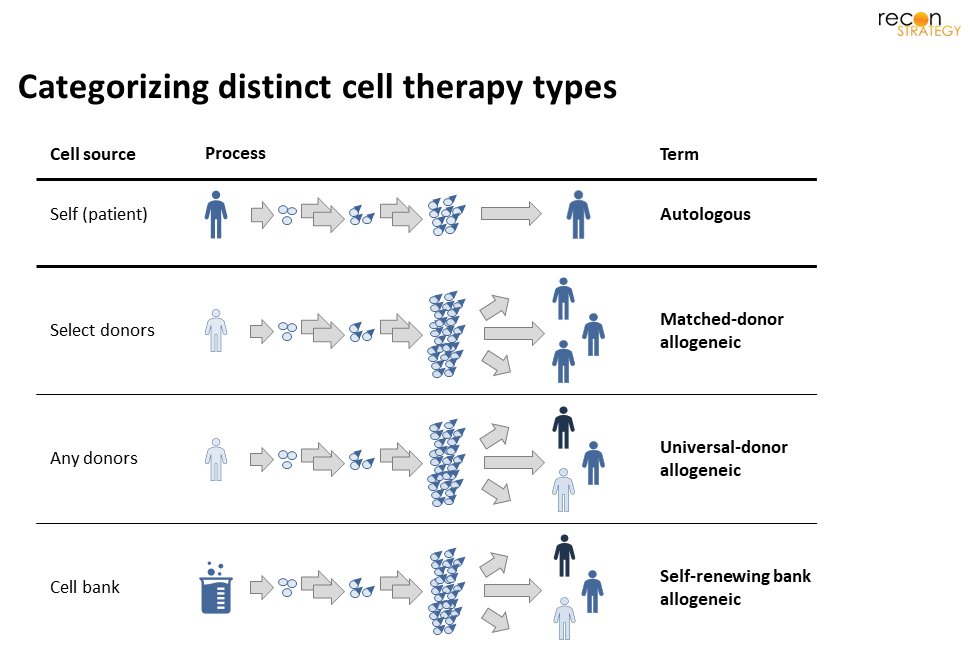
Before we begin, it is important to have a clear and consistent framework to discuss the various product types. It is critical to consider both the starting cell source and relation to intended recipients (Figure 2). This view highlights that beyond autologous, there are 3-distinct subtypes of allogeneic: matched-donor,[6] universal-donor, and self-renewing. For matched-donor, multiple donor ‘flavors’ are needed (akin to blood types); whereas a universal-donor product might be made starting with cells from any donor ‘flavor’. In self-renewing, a universal master cell bank (MCB) would replace human donations for all patients. (While self-renewing MCB could be made in multiple ‘flavors’, have seen little-to-no investment on this approach.)[7]
3. The state of cell therapy
It is important to highlight the nascent state of genetically-modified cell therapies today – less than a handful are on-market, and less than twenty thousand patients have received one of these products in the real world. That said, development is growing rapidly, as are the number of approved therapies. We provide a brief overview here.
Commercial assets
Today, all 7 approved cell therapies are autologous;[8] 6 of these products use CAR-Ts to targets hematologic cancer-antigens. Despite impressive comparative efficacy for relapsed/refractory cancers (5-year overall survival between 10-50% without cell therapy[9]), just 15k cancer patients have received a CAR-T to date.[10] In addition to being limited to 2nd/3rd line use, the viability of patients’ cells further narrows who has access to treatment. We delve into the specifics of why reliance on patient-cells precludes many in the “On-going challenges for autologous therapies” section. Only one commercial product targets a rare disease – bluebird bio’s Zynteglo (modified hematopoietic stem cells for beta thalassemia); despite struggling to gain traction in Europe, analysts expect US payers to cover treatment for ~1,500 patients (at $2.8M/patient).[11] No engineered allogeneic therapies are on-market today.[12]
Assets in clinical development[13]
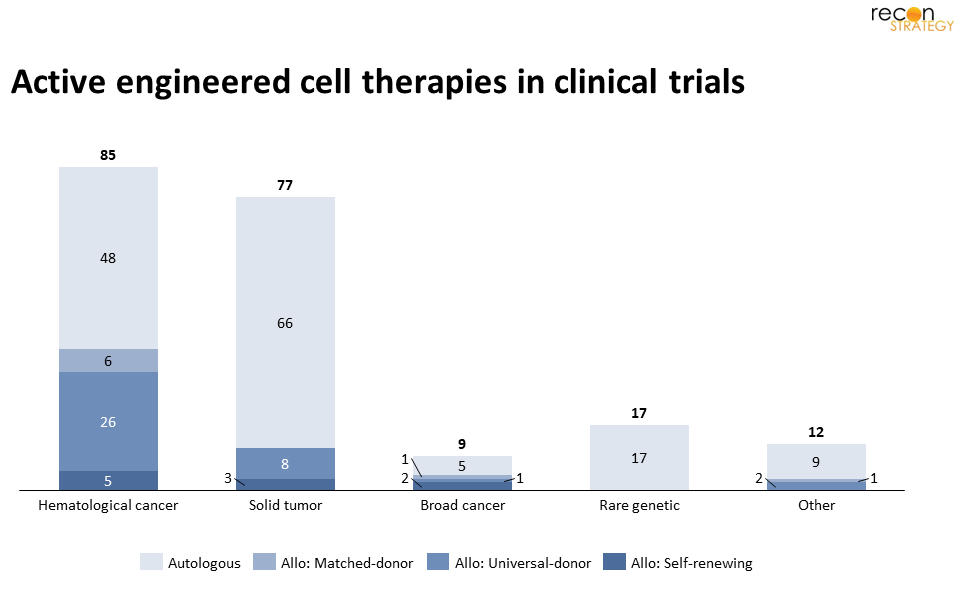
1. Autologous: Despite the growing interest in allogeneic, most clinical assets are autologous: 57% of early-stage, and all (7) late-stage assets. Many candidates target novel hematologic cancer-antigens, which might benefit relapsed patients (particularly if the tumor has lost previously targeted antigens). Despite few known solid-tumor specific antigens, developers are also evaluating autologous approaches (including T, NK, dendritic, stem and red-blood cells) for common cancers – often casting a “wide-net” across multiple tumor types. For example, Adaptimmune has pivotal trials for both esophageal and ovarian cancers with the same MAGE4-targeting T cell.[14] Outside of cancer, most late-stage products target rare disease blood disorders (e.g., sickle cell disease, Hemophilia A, beta thalassemia) via lentiviral modification of hematopoietic stem cells ex-vivo before re-engraftment.[15]
2. Matched-donor allogeneic: It is perhaps unsurprising to see just 7 matched-donor industry assets in development – given the inherent supply-chain complexity for multi-‘flavor’ product, vide supra. Typically, these assets are either supportive treatments for hematopoietic stem cell transplant patients (as immune reconstitution) or direct therapies that target cancers (e.g., CAR-VSTs[16]).
3. Universal-donor allogeneic: Most allogeneic assets fall in this category – albeit all remain pre-pivotal (i.e., none are Ph2/3 or Ph3). Nearly ⅔ (24 of 37) use genetically modified T cells, while others rely on NK or O-neg red-blood cells. Hematological cancers are the most common targets.[17] Some therapies (including Precision Biosciences’ lead candidate and Athenex’s CAR-NKT[18]) have positive interim Ph1 or Ph1/2 results, including for relapsed patients post autologous CAR-T treatment.
4. Self-renewing bank allogeneic: This group of assets are all in early-stage development for cancers. The two main types of MCBs used are (1) induced pluripotent stem-cells (iPSC) cells, which have been reverted to a stem-like state, and (2) immortalized lines descendent from spontaneously mutated donor cells, such as HeLa or NK-92. In this category, Fate Therapeutics is the largest player – their pipeline[19] relies on iPSCs and CAR-modification to create NK, T, and CD34+ cells.
4. On-going challenges for autologous therapies
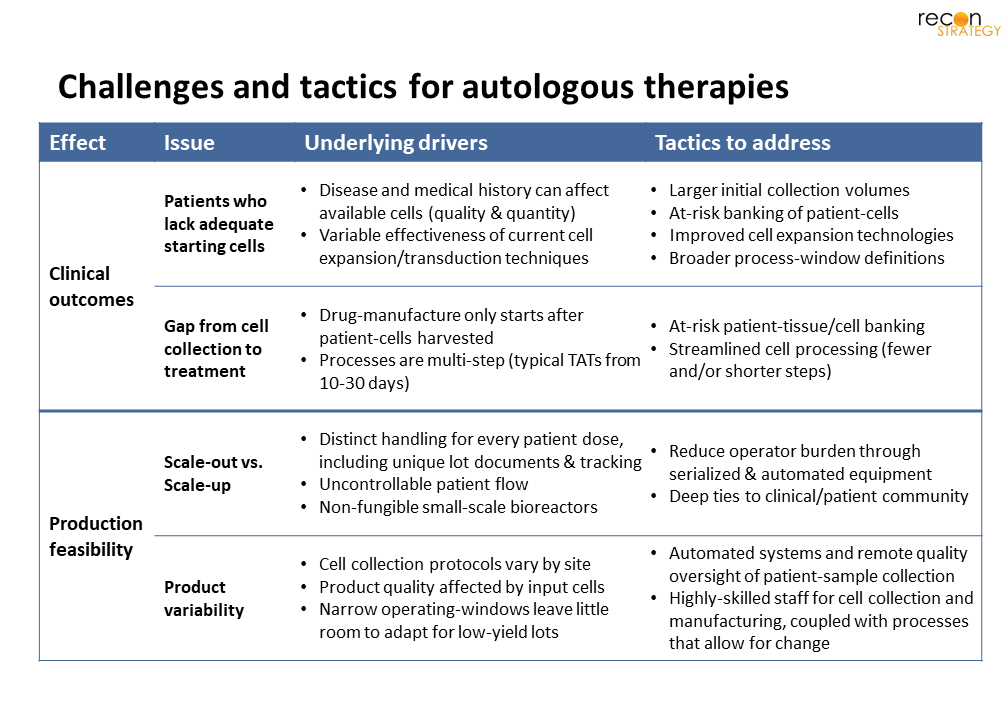
While commercial manufacturing has vastly improved since the early Kymriah rollout,[20] several issues remain. These issues affect both overall clinical impact and total cost-of-care. Below, we detail these challenges, along with promising approaches to minimize or eliminate them (Fig 4).
Clinical outcomes
Despite impressive results for some patients, many do not have access to these treatments. Patients may not be viables candidates due to inadequate cells or an inability to delay treatment. For aggressive cancers, it is not uncommon for both factors to contribute. The underlying drivers that create these hurdles, along with tactics to address include:
Patients who lack adequate starting cells: Medical history (including common first-line treatments) can negatively affect the quantity of cells salvageable from patient-tissues and blood, as well as their propensity for transduction/transfection or expansion. As a result, many patients’ cells simply cannot be converted to sufficient drug-product. The most obvious tactics to address this are (1) adjusting the treatment paradigm so these therapies are first-line, (2) at-risk banking,[21] or (3) collecting a greater volume of tissue.[22] Evidence in the form of randomized trials will be required for treatment naive-patients, while the latter approaches both increase patient burden, and add on ethical and regulatory considerations.[23] Improved cell expansion technologies, which reduce the necessary volume of starting patient-tissues, could be an alternative solution. New approaches under investigations
include lower cell density processes,[24] use of PI3K delta-inhibitors or VIP antagonists.[25]
Gap from cell-collection to treatment: Since product-manufacturing only begins once patient-cells are available, treatment delays can be substantial. Many patients cannot wait weeks to receive treatment, particularly when the disease is progressing rapidly. Current processes are multi-step (including cell isolation, activation, transduction, expansion and purification) and often require 1-to-3 weeks to complete.[26] Myriad efforts are on-going to adapt existing processes to minimize “vein-to-vein” time. Recent improvements include removal of the cell-expansion step (e.g., recently used for Novartis[27]) and streamlined LV-transduction with (as seen by Gracell’s FasTCAR[28]), each of which can reduce full CAR-T processing time to 1-2 days. Penn Medicine’s most recent 24-hour CAR T process even showed increased anti-leukemic activity in mice compared to standard processes.[29] However, since drug-preparation only begins after patient-cells are collected, treatment-delays cannot be fully eliminated.
Shorter vein-to-vein time would both reduce the cost of interim-care (often equivalent or greater than cell therapy cost3). However, these features would also expand the pool of addressable patients, resulting in even greater system cost. Earlier cell-banking (with extended storage periods) would exacerbate these costs further.
Production feasibility
Since each dose is unique, auto- cell products require bespoke manufacturing; typical economies of scale are out of reach. Additionally, greater operator expertise and oversight is needed to both (1) ensure adequate cell collection and (2) manage complex processing to drug-product. The factors leading to these issues, along with potential moderating tactics, are detailed below:
Scale-out vs. scale-up: Since each autologous product is unique to the patient, all doses are prepared individually, in separate reactors, with unique documentation and lot numbers. Adding complexity is the uncertainty of patient-flow, which makes production-scheduling extremely challenging. Lonza, Sartorius and Miltenyi offer tools (Cocoon, Ambr, and CliniMACS, respectively[30]) to automate and serialize (i.e., multiple units controlled in unison) reactors – which can reduce operator burden. In addition, while patient-flow cannot be controlled, an in-depth understanding of the patient-journey and strong ties to the relevant clinical community could improve visibility of expected product demand trends – enabling greater predictability and reactor scheduling.
Product variability: Consistency is a significant challenge for all bespoke manufacturing, and cell-therapy is no different. However, operating windows are defined on “normal” cells despite many patients’ cells not meeting “normal” cell specifications. Some companies have reduced the variability of patient-cells by taking on management and quality-oversight of cell-apheresis at hospitals.[31] Automated apheresis equipment could further improve quality control. Conversely, empowering operators to make lot-specific production choices (i.e., wide operating windows that allow for process variation), could make otherwise “out-of-spec” patient-cells viable. This latter tactic is only feasible with substantial change to regulatory guidance on manufacturing controls.[32] Further, it would increase the need for operator training and resources per drug-lot.
These cost challenges are a sticky issue. While automation and serialization can reduce the need for highly skilled operators, manufacturing remains bespoke – which is inherently prone to variability and leads to higher operator costs. Production schedules will continue to rely on patient-flow, and bioreactors will always be non-fungible to other products. As a result, while manufacturers will surely achieve lower-cost production through these tactics, autologous product COGs are likely to remain an outlier for biologic-manufacturing.
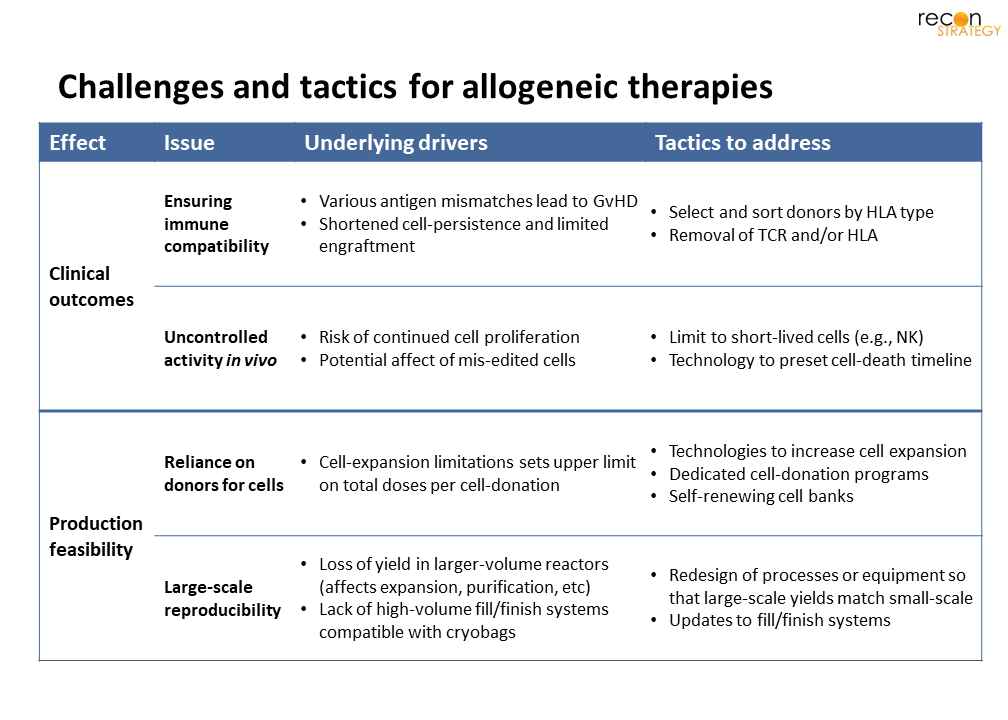
5. Hurdles allogeneic approaches need to overcome
Given the manufacturing challenges for autologous, it is unsurprising that investment continues on allogeneic therapies; these products would not suffer the risks of cell-deterioration,[33] treatment-delays, or bespoke manufacturing. However, fundamental uncertainties about the use of foreign cells (Figure 5) must be addressed before these products are brought to late clinical development.
Clinical outcomes
Since allogeneic products are prepared without patient-cells, patient-access is expected to be broader compared to autologous. With appropriate supply-chain management and demand-planning, all eligible patients could be treated without delay. But it’s not quite so easy as that – alien cells are more likely to be targeted and cleared by the immune system. While the cell-features the enable immune attack may be excised, fully anonymized cells present other risks. We detail the inherent challenges, and current approaches below.
Ensuring immune-compatibility: Mismatch of cell antigens (HLA-type) found on T- and B-cells leads to graft-vs-host disease (GvHD) and lower persistence of infused foreign cells. A traditional tactic is to select donors by HLA-type (and pooling donations by donor-type, for multi-donor approaches). However, multitude of “flavors” of every cell therapy, would be needed, given the range of HLA-types.[34] This complexity could be avoided by two alternative approaches: (1) use of GvHD-suppressive NK- or NKi-cells, or (2) removal of identifying receptors, such as TCR and HLA, to make CAR-T cells “universal” (UCART).[35] Firms with UCART approaches in the clinic include Cellectis and Allogene,[36] while Fate Therapeutics[19] leads the NK-approach.
Uncontrolled cell activity in vivo: The tactics that address matching and donor-reliance are double-edged. If cells lack TCR or HLA, key tools of the immune system are rendered impotent; making it even more critical that these cells’ actions are well-controlled. Regulators have closely watched investigational studies on all cell therapies for safety events, leading to a number of clinical holds.[37] Only one case was related to a chromosomal aberration (and eventually determined to have arisen in vivo [38]), the concern that even a small number of mis-engineered cells could lead to severe clinical consequences is real. It is perhaps unsurprising that many firms using self-renewing banks have focused on short-lived cells (e.g., NK, with an average lifespan of ~2-weeks). An alternative approach could be to engineer a “kill-switch” that sets a terminal limit to cell lifespan, or initiates apoptosis on exposure to select environments or antigens.
The above clinical uncertainties apply to all patients, not a mere subset. So, while the total opportunity is large (and uniform across patient-candidates), the risks are equally large and unknown. We hope that as programs progress from early- to late-clinical development, clear signals will emerge that confirms which approaches are viable – and which must be put aside.
Production feasibility
While a single large batch could provide multiple doses for any allogeneic product, experience with large-scale expansion of highly edited cells is scant. Several aspects of existing processes will need changes to ensure a robust and reliable supply of any cellular product. Critical features to address include:
Reliance on donors for starting cells: While a single donation can support multiple doses, continued cell-expansion increases the risk of differentiation and functional exhaustion. Consequently, a robust donor program (including extensive donor and sample screening) is critical for long-term supply most allogeneic products. However, a self-renewing MCB (akin to HeLa or NK-92[39]) would obviate the need for donors. Several well-funded biotechs are focused on this approach; examples include Fate Therapeutics, Century Therapeutics, Senti Bio and Garuda Therapeutics amongst others.[40]
Large-scale reproducibility: End-to-end, each step of cell-therapy production must be engineered to succeed at production-scale. Many techniques developed for small-scale (e.g., adherent-cell or co-culture supported expansion processes, cell separation via centrifugation) are less effective as scale increases – and have yet to be scaled to 100s of doses (let alone 1,000s) per batch. Additionally, while automated, cryovial-compatible fill/finish lines are available, a gap remains for cryo-bags, which are the typical format required for clinical doses. Suspension (in lieu of adherent) processes, which are amenable to large-volume stirred-tank-reactors, have already seen traction with both T cells and iPSCs.[41] We will need to see similar innovations that transfer purification and fill/finish processes to large scale as well.
While allogeneic products might circumvent many of the hurdles of autologous, much remains to be resolved. With sufficient resources and investment, we expect bioprocess-engineering can identify solutions to large-scale reproducibility, but the question of how additional cell-modifications impact safety and efficacy can only be answered through clinical evaluation. Although a more distant goal, we believe the potential of self-renewing banks to simplify supply chains (and excise donor program costs) will keep investor interest high.
6. Concluding thoughts
Although autologous therapies, as a class, are more advanced, there are reasons to believe allogeneic could eventually dominate, particularly for cancer indications. These products are inaccessible for many – particularly if lymphopenic (due to prior-treatment) or if PBMCs are rich in circulating tumor cells. Even if the treatments move to first line, the risk of progression during therapy-preparation remains a significant concern for aggressive cancers. In contrast, an off-the-shelf, universal allogeneic product could serve all patients, regardless of clinical history. For cancers where clinical value is measured in months of survival, the risk of unintended genetic changes seems justified. Not surprisingly, preclinical momentum has shifted to allogeneic (new program starts outnumbered autologous since 2020).[42] This trend is likely to continue until a negative safety signal arises that justifies abandoning the approach.
In contrast, for rare-disease, where annual treatment numbers are low (after the initial existing-patient bolus), the complexity of bespoke manufacturing may remain justified. These diseases are often diagnosed early-in-life, without any cell-depleting treatment-history – and thus fewer relevant patients would be precluded from treatment. Further, since many more quality adjusted life years (QALYs) are gained for rare diseases than cancer, these treatments are more likely to be viewed as cost effective, even when >>$1M/patient.[43] Finally, most rare-disease patients could enjoy otherwise healthy lives post-treatment; the lifetime tumorigenicity risk from an allogeneic product is inherently greater. Although the lower cost of a bulk-produced drug remains desirable, the risk of future malignancy may be a concern – at least until the category has an established safety record.
Sarah Dolman, PhD
Partner
Maggie Pickard
Associate Consultant
[1] We reserve the term “gene therapy” for in vivo gene-editing products such as Zolgensma or Luxturna. In the context of this paper, we exclude non-modified cell therapies, (any therapy that does not undergo genetic engineering). Many unmodified stem cells or virus-specific T cells (VSTs) are not included.
[2] Acute lymphoblastic leukemia, Novartis Press Release, June 2022
[3] (a) Sahli, “Cost management for sky-high, high-cost gene therapy.” Prime News, Aug 2021. (b) Minemyer, “The gene therapy pipeline may be at a ‘tipping point’. Here’s why Optum says payers need to take notice.” FierceHealthcare, Aug 2022
[4] According to SEER, the annual age-adjusted incidence across all leukemias, lymphomas and myelomas (excluding Hodgkin’s lymphoma and CLL) is ~100k patients. Seer Incidence Statistics, 2022
[5] PharmaProjects data as of August 7, 2022. Includes all cell therapy products identified as using allogeneic or autologous cells. Only includes US-based therapies. Does not take into account non-industry therapies. Products entering early clinical phase are either entering Ph1 or Ph1/2. In 2022, 53 out of 63 autologous and 64 out of 86 allogeneic therapies entering preclinical phase appear genetically engineered (3 autologous, 2 allogeneic unclear). 19 of 30 autologous and 19 of 34 allogeneic therapies entering early clinical phase appear genetically engineered.
[6] Matched-donor therapies may be prepared specifically for an individual patient, however cell-expansion technologies can usually create multiple-doses from a single-donation.
[7] While it may be feasible to create self-renewing ‘flavored’ cells, we have only seen efforts toward universal self-renewing cells.
[8] As of August 2022, these include Kymriah, Abecma, Yescarta, Tecartus, Carvykti, Breyanzi and Zynteglo.
[9] (a) ALL Haematologica, 2010(b) DLBCL Blood, 2017 (c) FL J Clin Oncol, 2015 (d) MM Ann Hematol., 2021
[10] (a) Over 6,800 patients have received Yescarta, Mar 2022 (b) 5,300 treated with Kymriah Aug 2021(c) Tecartus quarterly revenue, Gilead (d) Abecma and Breyanzi quarterly revenue, BMS
[11] (a) Autologous cells, modified to produce a functional hemoglobin. FDA Press Release, August 17, 2022 (b) FiercePharma, August 17, 2022
[12] Examples of non-engineered allogeneic treatments on-market today include stem-cell transplants and lympho-cyte infusions. These products are typically used as supportive therapy post-transplant or immuno-depletion.
[13] PharmaProjects data as of August 7, 2022. Only includes immunological cell therapy products that were identified to be genetically engineered. Only includes US-based therapies and does not include non-industry assets.
[14] Adaptimmune Pipeline 2022
[15]Kohn, Pai, Sadelain, “Gene therapy through autologous transplantation of gene-modified hematopoietic stem cells.” Biol Blood Marrow Transplant., 2013
[16] 7 non-engineered virus-specific T cells (VSTs) in trials are not included in our landscape review. There are also several academic-led early trials of engineered VSTs.
[17] Haslauer, Greil, Zaborsky, et al “CAR T-Cell Therapy in Hematological Malignancies”. Int J Mol Sci., 2021.
[18] (a) Precision Biosciences PBCAR0191 Press Release June 2022. (b) Athenex KUR502 Press Release, April 2022.
[19] Fate therapeutics pipeline, Aug 2022
[21] Palen, Zurko, Johnson, et. al, “Manufacturing CAR T cells from cryopreserved peripheral blood cells: time for a collect-and-freeze model?” Cytotherapy, 2021
[22] Jalowiec, Pabst, Bocksrucker et. al. “How to Collect the Minimum-Targeted CD3+ Cells for CAR-T Therapy- the Bern Approach” Blood, 2019
[23] Levine, Munsie, Levine et. al “The peril of the promise of speculative cell banking: Statement from the ISCT Committee on the Ethics of Cell and Gene Therapy” Cytotherapy, 2022
[24] Stemcell Technologies, Oct 2018.
[25] Petersen, Hassan, Morris et. al. “Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists”, Immunobiology and Immunotherapy, 2018.
[26] For example, Gilead reports the real-world turnaround for Yescarta is, on average, 17-days; while BMS targets 24-days for Breyanzi (with a minimum expectation of 7-days), per FiercePharma, Dec 2021.
[27] Engels, Zhu, Yang, et al, “Preservation of T-Cell Stemness with a Novel Expansionless CAR-T Manufacturing Process, Which Reduces Manufacturing Time to Less Than Two Days, Drives Enhanced CAR-T Cell Efficacy”, Blood, 2021
[28] Yang, Jiang, Zhang, et al, “Successful 24-Hours Manufacture of Anti-CD19/CD22 Dual Chimeric Antigen Receptor (CAR) T Cell Therapy for B-Cell Acute Lymphoblastic Leukemia (B-ALL)”, Blood, 2020
[29] Ghassemi, S., Durgin, J.S., Nunez-Cruz, S. et al. “Rapid manufacturing of non-activated potent CAR T cells.” Nat Biomed Eng, 2022
[30] (a) Lonza, Cocoon Brochure, 2022 (b) Sartorius, Ambr Multi-Parallel Technology, 2022, (c) Miltenyi Biotec, Cell Manufacturing Platform, 2022
[31]Chapman, “Overcoming the Quality Challenges of a CAR-T Cell Product: One Company’s Experience”, Redica Systems, 2020.
[32] (a) FDA “Human Gene Therapy Products Incorporating Human Genome Editing; Draft Guidance for Industry”, FDA Guidance, March 2022 (b) FDA “Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products; Draft Guidance for Industry”, FDA Guidance, March 2022
[33]Caldwell KJ, Gottschalk S and Talleur AC. Allogeneic CAR Cell Therapy—More Than a Pipe Dream. Front. Immunol, 2021
[34] HLA genes code for receptors on most nucleated cells, with multiple types across both MHC1 and MHC2. Major MHC1 include HLA-A, HLA-B & HLA-C, minor MHC1 are HLA-E, HLA-F & HLA-G. MHC2 types include HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA, amongst others.
[35] Zhao, Lin, Song & Liu “Universal CARs, universal T cells, and universal CAR T cells” J of Hematology & Oncology, 2018.
[36] Cellectis clinical pipeline, Allogene research pipeline.
[37] (a) Celyad clinical hold lifted, Aug 2022, (b) Cellectis clinical hold, Jul 2020, (c) Bellicum clinical hold, Jan 2018
[38] Allogene clinical hold lifted, Jan 2022. It is worth noting that a comparable amount of clinical holds have occurred amongst autologous therapies; in the past year alone, Tmunity Therapeutics and Atara Biotherapeutics have paused clinical trials of autologous therapies due to patient deaths –Tmunity FDA hold, Atara Bio clinical pause
[40] (a) Century Therapeutics, (b) Fate Therapeutics, (c) Senti Bio (d) Garuda Therapeutics
[41] (a) Gatla, Uth, Levinson et al “Enabling Allogeneic T Cell-Based Therapies: Scalable Stirred-Tank Bioreactor Mediated Manufacturing” Front. Med. Technol., 2022 (b) Pigeau, Caesar, Dulgar-Tulloch, “Commercial Scale Manufacturing of Allogeneic Cell Therapy”, Front. Med., August 2018
[42] Preclinical allogeneic program starts in 2020, 2021, and 2022 were 69, 108, and 86, respectively, compared to 51, 99, and 63 for preclinical autologous programs.
[43] As an example, ICER’s analysis determined Zyntelgo as cost-effective, even at $3M (beyond bluebirdbio’s intended price of $2.8M). Endpoints News, Aug 2022