
Introduction
In 2017, the FDA approved Luxturna for the treatment of Leber’s congenital amaurosis (LCA), marking the first gene therapy used for an inherited disease in the U.S. The curative potential of gene therapy garnered significant pharma industry interest over the next few years, with notable multi-billion dollar acquisitions from Novartis and Roche. Since that initial flurry of activity, there has been a meaningful pull-back, with pharma industry investment in cell and gene therapy declining from $22.7B in 2021 to $11B in 2023, according to JP Morgan estimates. Specific to in-vivo gene therapy, the waning interest has been attributed to a myriad of drivers. Challenges in market access and reimbursement have been one concern, primarily due to the high prices owing to the nature of one-and-done treatments, a topic we explored several years ago. Concerns with safety in AAV gene therapy is another commonly raised issue, with alternative delivery approaches a potential future solution. Perhaps most important has been the mixed commercial success of in-vivo gene therapies to date, with multiple market withdrawals in Europe and generally underwhelming revenues accrued in the U.S. In this piece, we explore the current commercial performance of the AAV-based gene therapies approved in the U.S. by 2023, to better understand the market realities and identify key learnings for future products.
Underwhelming commercial performance of Luxturna potentially skewed by overestimated patient population
Luxturna is the only approved therapy for LCA and has captured the wide majority of eligible patients
Spark Therapeutics’s approval of Luxturna in 2017 was a milestone for gene therapy in the U.S., leading to its subsequent acquisition by Roche. Luxturna is indicated for patients with biallelic RPE65-associated retinal dystrophy, having either retinitis pigmentosa (RP) or Leber congenital amaurosis (LCA) and sufficient viable retinal cells as determined by the treating physician. Around the time of launch, Wall Street analysts were bullish on commercial expectations with peak sales ranging between $350M and $750M by 2022, and total lifetime revenues of >$1-3B. In reality, Luxturna’s total sales from 2018 through 2023 have been closer to $250M. Interestingly, KOLs have reported most patients eligible for Luxturna have been treated, indicating significant patient uptake of Luxturna. So why the discrepancy?
In speaking with KOLs, it appears the discrepancy is related to patient population sizing issues. At the time of launch, the medical literature estimated 1,000 to 2,000 patients in the U.S. with biallelic RPE65-associated retinal dystrophy, with relatively even splits between RP and LCA patients. RP affects approximately 80,000 individuals in the U.S. and an estimated 1% of RP cases were believed to be attributable to RPE65 mutations, thus treatable by Luxturna. However, the 1% figure is largely based on a handful of small, targeted registry studies. In reality, KOLs report those treated with Luxturna are predominantly LCA patients, and the actual total number of U.S. patients with biallelic RPE65-associated retinal dystrophy is closer to 650. It appears past epidemiology studies exploring RP cases attributable to RPE65 mutations (1%) may have led to an overestimation, thus significantly skewing the total number of eligible patients and Luxturna’s market potential. With few to no RP patients being treated with Luxturna, consensus estimates appear to have overextrapolated from small studies with low reliability and limited generalizability, a phenomenon that we see in the rare disease space.
Thus, despite not appearing a financial success relative to commercial expectations, the treatment of nearly all eligible patients in a relatively small population still signifies a market success. The perceived limited commercial performance of Luxturna appears to be driven by incorrect expectations set by the market, as opposed to lack of patient uptake. Hence, as long as companies are clear with investors and themselves about the limited financial potential of ultra-rare diseases and plan R&D investments accordingly, the Luxturna experience suggests patients with significant unmet need in these conditions will use novel treatments.
Zolgensma thrives with newly diagnosed SMA patients, leveraging competitive efficacy and long-term data
In May 2019, Zolgensma was approved by the FDA for treating children under 2 years of age with spinal muscle atrophy (SMA). SMA affects 10,000-25,000 children and adults in the U.S., and 3 treatments are now currently FDA approved: Spinraza (antisense oligonucleotides (ASOs)), Zolgensma, and Evrysdi (small molecule). In 2020, Zolgensma sales totaled over $920M globally and steadily grew to $1.2B in 2023. In 2023, all 3 SMA treatments reached blockbuster status, with Zolgensma lagging with the lowest, and declining, total sales (Figure 1).
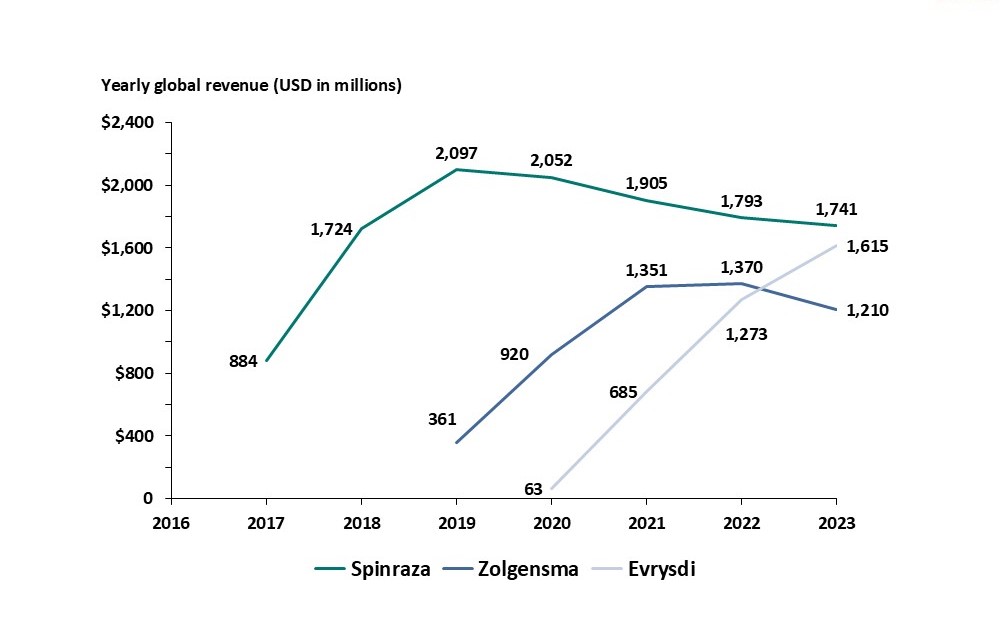 Figure 1, Company financial filings
Figure 1, Company financial filings
So while Zolgensma has clearly been a greater financial success as compared to Luxturna, does the relatively lower commercial performance suggest headwinds for AAV treatments compared to their non-gene therapy counterparts? A deeper analysis suggests not in this case.
SMA refers to a group of hereditary conditions that vary in age of presentation and severity, but all affect motor neurons. Type 1, or infantile-onset SMA, typically presents before 6 months of age and involves severe muscle weakness and trouble breathing, walking, and many children historically did not survive beyond age 2. Other forms of SMA, specifically type 2 through 3, affect children at later time points, with variable levels of muscle impairment and impacts on life expectancy. While Spinraza and Evrysdi are indicated for all three types of SMA, Zolgensma is indicated for the most severe patients, specifically in type 1 or 2 patients that present under 2 years old. In this context, Novartis has stated Zolgensma was primarily treating newly diagnosed patients in its first year after approval, claiming ⅔ of newly diagnosed patients in the U.S. were being treated with Zolgensma. Recent analyses show Zolgensma was the treatment of choice in newly diagnosed patients, growing to approximately 80% of U.S. share, according to data from Cure SMA (Figure 2). Zolgensma’s apparent lag in overall sales behind Spinraza and Evrysdi obscures its dominant uptake in its indicated, newly diagnosed population.
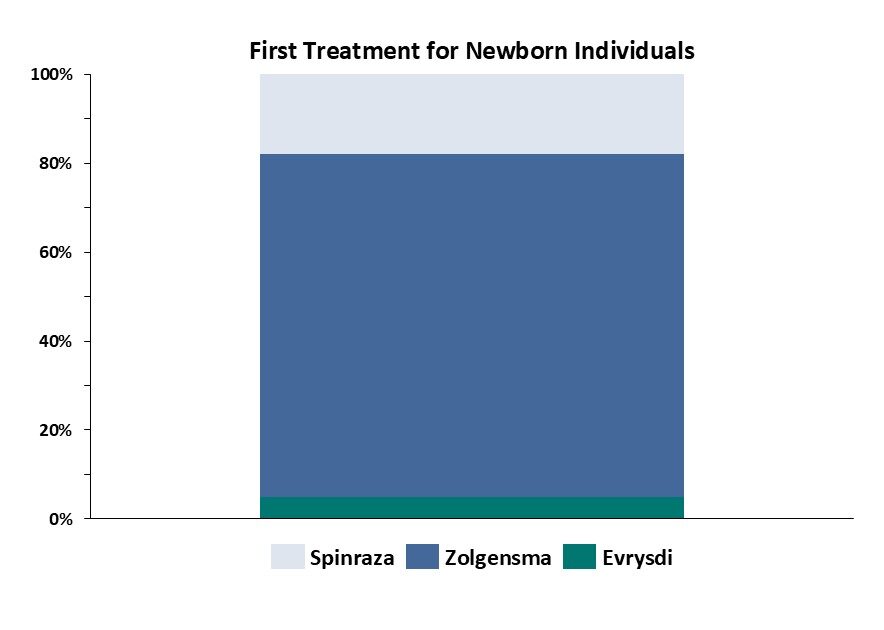 Figure 2, 2022 Cure SMA newborn screening dataset across 42 U.S. states
Figure 2, 2022 Cure SMA newborn screening dataset across 42 U.S. states
Why has Zolgensma been the treatment of choice for these patients? It appears that it is due to a mix of trial data and mechanism of action. While all three approved treatments demonstrated significant efficacy in their respective pivotal trials, KOL interviews suggest an efficacy advantage to Zolgensma based on somewhat stronger data across its pivotal trials (STR1VE and SPR1NT) and sustained survival and muscle function improvement over a longer term compared to the other 2 treatments (LT-002). Interviews with KOLs also note that Zolgensma targets the SMN1 gene mutation directly, in contrast to Evrysdi and Spinraza which increase SMN2 protein levels to indirectly address the SMN1 deficiency– with the former being preferred by clinicians. Zolgensma therefore is a case demonstrating strong financial and market performance is possible: a sufficiently large patient population with significant unmet needs, coupled with meaningful differentiation from competitors can lead to a clear commercial success.
Roctavian and Hemgenix off to slow starts in an established Hemophilia market
Roctavian’s durability concerns present challenges in competing with existing therapeutic alternatives in Hemophilia A
BioMarin’s Roctavian became the first gene therapy approved by the FDA for treatment of Hemophilia A in June 2023. Prior to its commercial launch, consensus estimates predicted $1.8B peak sales by 2026, representing ~5% patient share. In 2023, Roctavian generated $3.5M in sales, far below BioMarin’s own estimated range of $50M-$150M. In response to the underwhelming commercial performance, BioMarin announced it would be narrowing Roctavian’s focus to just 3 countries: U.S., Germany, and Italy, with the hope to become profitable by 2025. Why was Roctavian’s commercial performance so different from the Zolgensma experience above?
Hemophilia A is a crowded market, with more than ten prophylactic treatments currently approved. Most current treatments have demonstrated clinically meaningful efficacy (annual bleed rate (ABR) < 1.5) and years of safety and durability data. Hemlibra, approved in 2017, now commands over 40% of this market, driven largely by its differentiated route of administration and less frequent dosing regimen: whereas preceding factor replacement therapies are typically dosed weekly, Hemlibra is a biweekly subcutaneous treatment, and is sometimes required even less frequently based on patient response. This existing arsenal of highly efficacious and durable treatments left less room for Roctavian, given a much more modest unmet need primarily based on reducing treatment burden. In BioMarin’s Phase 3 GENEr8-1 study, Roctavian showed reasonable efficacy data in mean ABR (2.6), relative to Hemlibra (1.3) and other approved therapies (ALTUVIIIO; 0.70). As an AAV gene therapy, Roctavian potentially differentiates itself from alternative therapeutics with its promise as a one-and-done treatment. However, durability has been an issue: Factor VIII activity levels for patients treated with Roctavian were shown to fall off after 12 to 18 months of treatment in patients in the Phase 3 GENEr8-1 study, and these concerns lead the FDA to initially not approve Roctavian in 2020. Now approved but still with some doubts on durability, KOLs have reported most patients unlikely to switch from current treatment to Roctavian, citing questionable durability as the main issue (Figures 3 and 4). Given that the primary advantage of Roctavian is a curative treatment that eliminates the treatment burden, issues with durability have proven to be a defining driver in its lack of success.
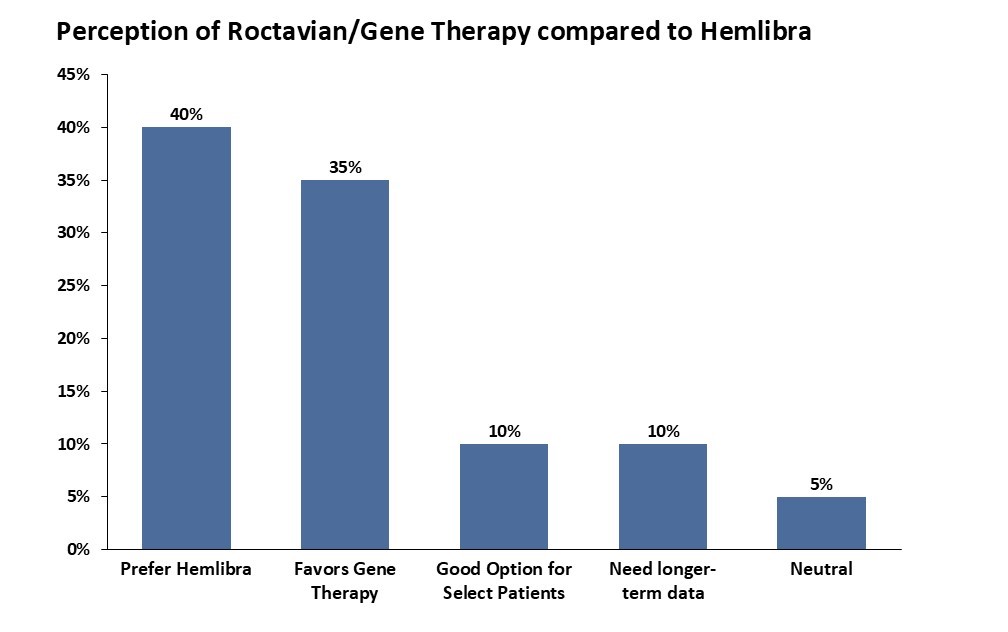 Figure 3, JP Morgan 2022
Figure 3, JP Morgan 2022
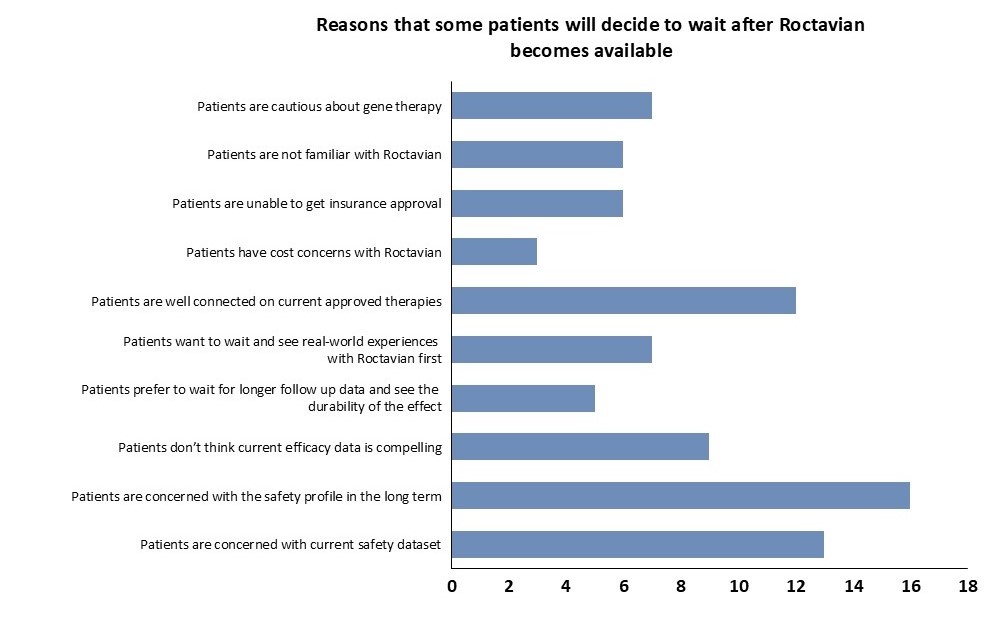 Figure 4, Jeffries 2022
Figure 4, Jeffries 2022
Hemgenix off to a slow start, but Hemophilia B presents a more manageable opportunity
The market in Hemophilia B is a bit less crowded, with 6 prophylactic factor replacement therapies ranging from 1-3 doses per week, and one weekly subcutaneous therapy (Hympavzi) that was just approved October 2024. CSL Behring’s gene therapy Hemgenix was approved by the FDA in November 2022. However, similar to Roctavian, Hemgenix uptake has fallen below expectations, being used in less than 20 patients through August 2024.
Compared to market leaders Idelvion and Alprolix (mean ABRs 3.4 and 3.0, respectively), Hemgenix compares favorably: Patients treated for 7-18 months showed competitive efficacy in mean ABR (1.51). Yet similar to Roctavian, analysts have reported providers are reluctant to switch from standard factor replacement therapies due to durability concerns. To its credit, CSL is addressing this over time, releasing some durability data last year from a follow-up of the pivotal trial showing nearly identical mean ABR (1.52) and stable FIX activity levels through 3 years post-treatment, contrasting with Roctavian in Hemophilia A. As this data becomes increasingly available and shared with the Hemophilia community, a recent KOL survey by Wells Fargo has reported growing interest from patients in learning more about Hemgenix, citing durability as a key factor. Additionally, CSL has credited its slow uptake to reimbursement and market access challenges, consistent with key clinicians and specialty pharma heads who have highlighted lags in contracting for gene therapy treatments. Given the uncertain market dynamics and a new subcutaneous competitor in Hympavzi, albeit one that is still a weekly treatment, it remains to be seen how the market will respond.
The Hemophilia experience in gene therapy offers a cautionary tale. The unmet need for Hemophilia A and B patients is much more modest compared to LCA and SMA given the efficacy afforded by the existing standard-of-care. As important, neither Roctavian nor Hemgenix had sufficient data at launch to compellingly address its core value proposition as a one-and-done curative treatment. Perhaps over time, continued follow-up with clinical trial patients and real-world evidence can provide enough durability data to change the minds of treating physicians and patients. At the same time, the slow start and initial negative brand perception associated with the lack of sufficient durability may be too difficult to overcome, which may explain Pfizer’s recent decision to end collaboration with Sangamo’s Hemophilia A gene therapy.
Elevidys on the way to strong commercial performance in a DMD market with significant unmet need
Duchenne muscular dystrophy (DMD), a severe muscle degenerative disease caused by mutations in the dystrophin gene, affects 1 in 3,500 to 5,000 males born worldwide. Most patients, predominantly young boys, lose their ability to walk by 12 years old and have an average life expectancy of 28 years. Previously approved therapies, including corticosteroids and exon-skippers, have very modest clinical effects, but given the severity of unmet need and a very proactive patient community, these therapies have shown meaningful uptake. However, it is very clear to both patients and clinicians that a significant unmet need remains. In June 2023, Sarepta’s Elevidys received accelerated approval by the FDA for treating DMD in ambulatory individuals between 4 and 5 years of age, and in its first 12 months post-launch, Elevidys totaled $455M in U.S. sales. Recently having received a broader label for all DMD patients ages 4 and older, consensus estimates peak sales over $3B by 2030, opening the door for Elevidys to become the most commercially successful gene therapy to-date. What is different about Elevidys that puts it on a multi-billion dollar commercial path?
Contrasting with the currently approved therapies for DMD, Elevidys offers a meaningfully differentiated product by leveraging its impact on biomarkers, clinical outcomes, and safety. Clinical trials show Elevidys-treated patients reaching ~40-55% of normal dystrophin expression levels, a protein that plays a critical role in maintaining muscle function and integrity. In addition, the EMBARK pivotal trial showed statistically significant effects on several timed-function tests such as Time to rise (TTR) and 10-meter walk, relative to placebo. With safety concerns surrounding other treatments in the pipeline, including Pfizer’s highly anticipated gene therapy, Elevidys has an advantage against potential future competitors as well. Although having missed its primary endpoint in the EMBARK trial, Elevidys has compellingly differentiated itself from the market with its effect on microdystrophin levels, impact on several clinical function tests, and good safety experience to date. In a disease marked with tremendous unmet need and an active group of patients, Elevidys is able to bring significant value to patients, based on this data, thus paving a path to becoming one of the most commercially successful gene therapies.
Final thoughts
Despite the excitement around the promise of curative treatments with one-and-done potential, the experience so far among AAV-based in-vivo gene therapies suggests a relatively banal lesson. The business fundamentals that are key to commercial success in biopharma are just as important in gene therapy: thoroughly evaluating epidemiology, robustly assessing unmet need to ensure a sufficient addressable patient population, ensuring product profiles are targeted towards sufficient competitive differentiation and developing data packages with market-relevant efficacy and safety information. There is no doubt that there are particular commercialization challenges with one-time therapies and their associated high prices to date, and the outcomes based agreements such as the one recently announced by CMS, Vertex and Bluebirdbio are examples of the innovative elements needed to make these therapies easier for our health care system to absorb. However, the U.S. experience to date with gene therapies suggests that commercial success is most tied to known success factors in pharma, and despite the buzz associated with gene therapy and newer therapeutic approaches on the horizon, companies will be best positioned by ensuring they take a tried-and-true, calculated approach for investing in these areas.