
Introduction
Today’s advanced therapies target devastating diseases with high unmet need. Given the urgent medical need (and pressure from patient advocates), regulators increasingly must make decisions based on early, incomplete data when it comes to durability, real-world effectiveness, and cost-effectiveness – all while bearing costs that can exceed $1 million per patient.[1]
Policymakers globally are grappling with how health systems should manage this combination of high upfront costs and uncertain long-term value. In the United States and Europe, debates have intensified around pricing frameworks ranging from outcomes-based reimbursement to international reference pricing and “most-favored nation” (MFN) benchmarking. At the same time, the UK’s pharmaceutical pricing framework has faced international debate. Critics have argued that strict cost-effectiveness thresholds are merely a mechanism for suppressing drug prices, while proponents of the UK’s approach view the system as a necessary tool for managing finite healthcare budgets. Both vantage points reflect a shared concern: how to ensure access to transformative therapies while maintaining sustainable healthcare spending.
Outcomes based pricing has been one of the ways to tackle this uncertainty, where a proportion of the price is held until the outcome is clear. Theoretically, the UK’s healthcare system is well-suited for such an arrangement: as a single, government-funded system covering patients for life, the National Health Service (NHS) can capture the full long-term economic benefit of upfront investments in high-cost treatments and cures. Outcome confirmation should be accessible: the NHS’s consolidated care delivery model allows for more consistent and timely tracking of patient outcomes. And there are low risks for incentive misalignments: the NHS makes no money on medicines distribution and has a dedicated pathway for evaluating medicines that avoids conflicting incentives (which afflict the US market).
Yet formal OBAs are not the model the UK uses. While the UK did initiate some classic OBAs for Velcade[2] and Votrient[3] in the early 2000s, increasingly they have evolved their system to a managed access structure. How it has navigated the uncertainty and promise of novel therapies offers valuable lessons for other markets, including the US. In this analysis, we will examine how much OBA benefit the NHS actually captures through its Managed Access program.
Overview of Managed Access
The NHS delegates assessment of new treatments to the National Institute for Health and Care Excellence (NICE), which analyzes the value of innovative, high-impact, or potentially controversial technologies using a multi-stakeholder deliberative process. When NICE recommends a treatment, NHS commissioners must fund it across England.[4] The National Institute for Health Research Innovation Observatory, an academic group at Newcastle University, conducts “horizon scans” and notifies NICE about emerging technologies ~20 months before marketing authorization. All new cancer drugs and major new licensed indications are automatically referred to NICE by this group. Appraisals determine whether a drug is approved for routine access, managed access, or rejected.
Historically, NICE generally applies an incremental cost-effectiveness threshold (ICER) of £20,000 –£30,000 per quality-adjusted life year (QALY) through its Single Technology Appraisal (STA) program.[5] For rare and severe diseases, however, NICE has relaxed the ICER threshold:
- Disease severity: Since 2022[6], for severe conditions (e.g., cancer), ICER thresholds can be increased to £36–51K per QALY under NICE’s “severity modifier”.[7],[8]
- Disease prevalence: For ultra-rare conditions, the Highly Specialized Technologies (HST) program[9] allows thresholds up to £300,000 per QALY.[10],[11]
While relaxed QALY thresholds can help address devastating unmet patient need, they do not solve the uncertain data problem. Accordingly, NICE introduced the Managed Access Agreements (MAAs) model in 2016. These allow early controlled prescribing and reimbursement while additional evidence is gathered. Financial exposure is managed by limiting reimbursement source to two capped funds – the Cancer Drugs Fund (CDF) and the Innovative Medicines Fund (IMF) – each with an annual budget of £340 million, together accounting for about 3.5% of England’s £19 billion drug spend.[12] Patients receive treatments through the dedicated funds, biopharmas collect real-world data to confirm long-term value, and after several years, the therapy is reappraised.
This approach broadly captures many of the potential advantages of OBAs while avoiding some of the complexities and burdens of OBAs for individual medication/patient combinations. It does this by, essentially, conducting one large-scale OBA where:
- Upfront access before outcome is verified: Patients most likely to benefit from the medicines gain access with consistent national coverage and capped NHS spending
- Transparent process for verification: NICE can confirm biopharma’s QALY claims
- Payoff for biopharma if outcomes are verified: If results confirm expectations, biopharma can secure broader access beyond the MAA funds and potentially negotiate an adjusted price.
The following case studies will explore how these mechanisms have worked in practice.
MAA for cancer
Since its creation, NICE has reviewed 374 cancer drug indications, of which 55 received funding through the CDF at one point in time. Currently, around 80% of oncology drugs appraised by NICE have received positive recommendations (routine or managed access).[13] Of the 26 oncology drugs spanning 55 indications that entered the CDF between 2016 and 2022, 21 had within-indication follow-on results. Most moved to either full recommendation (10 of 19) or an optimized listing (7 of 19) (Appendix 1).
Upon reappraisal with mature data, CDF drugs typically follow one of three pathways (Exhibit 1):
- Routine commissioning, if follow-up confirms clinical benefit and cost-effectiveness
- Optimized recommendation, if the benefit is clear only for a specific subgroup
- Not recommended, if evidence fails to confirm meaningful benefit or acceptable cost-effectiveness
Exhibit 1. CDF cancer drug evaluation pathway
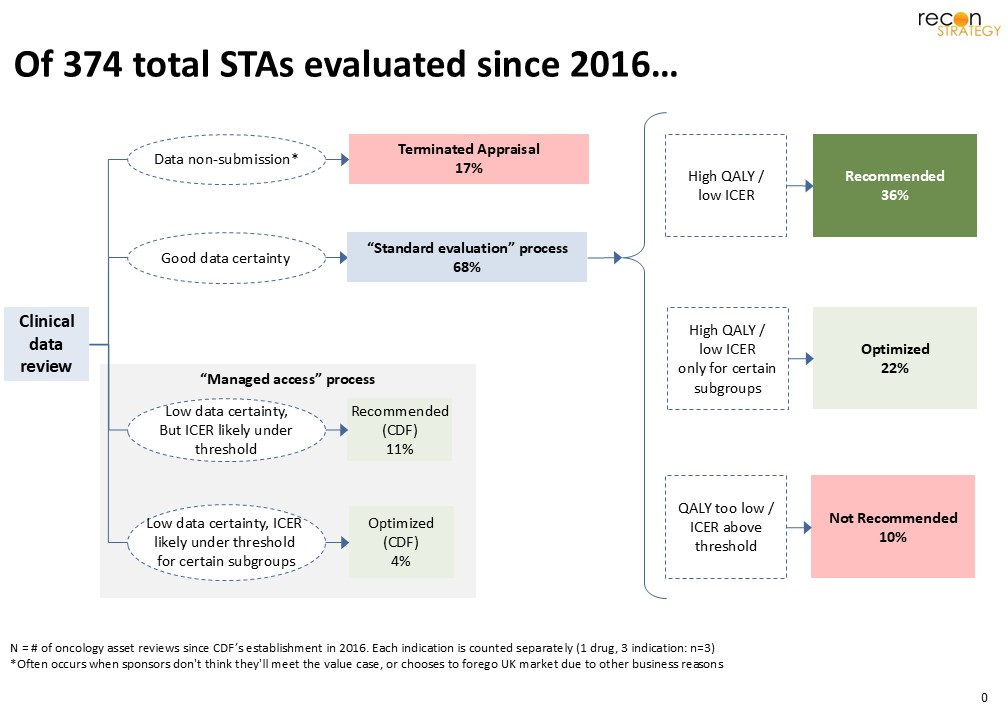
Below are several cases that demonstrate how this model works in practice.
Recommend (CDF) → Optimized / Recommend
Crizotinib (ROS1-positive NSCLC)
- First CDF entry (TA529, 2018): Entered the CDF based on PROFILE 1001, a single-arm phase I study (n=53) with mostly pre-treated patients (n=46). Overall survival (OS) data were deemed immature[14], and the absence of a comparator arm created uncertainty in cost-effectiveness modelling. These uncertainties, coupled with the high unmet need of ROS1-positive NSCLC, led NICE to grant CDF entry rather than full recommendation.
- Reappraisal data (2024): Included additional 4 years of OS data from PROFILE 1001, plus indirect comparisons with entrectinib (now licensed for the same indication, head-to-head data expected 2027). The reanalysis found no statistically significant OS difference between crizotinib and entrectinib, and incremental cost-effectiveness ratios (ICERs) were similar.
- Result: Fully recommended for NHS use, but only for patients who have not previously received a ROS1 inhibitor (including entrectinib).
Recommend (CDF) → Optimized
Pembrolizumab + carboplatin + paclitaxel (untreated metastatic squamous NSCLC)
- First CDF entry (TA600, 2019): Entered the CDF based on interim KEYNOTE-407 data, which suggested varying but positive OS and PFS benefits across all PD-L1 subgroups, but immature within-group data. At entry, there was no PD-L1 criteria for the combination therapy (PD-L1 thresholds were used only for pembrolizumab monotherapy). Cost-effectiveness estimates were uncertain for subgroups of varying PD-L1 levels, but ICER was low enough for MAA.
- Final KEYNOTE‑407 data (TA770, 2021) demonstrate that the survival benefit of Pembro + chemo in patients who had PD-L1 >50% was not statistically significant. Final scope was limited to PD-L1 1–49%, or ≥50% if urgent intervention is required.
- Result: NICE issued an optimized recommendation—restricting combination use to PD-L1 <49%, or PD-L1 ≥50% only when rapid clinical intervention is required.
Recommend (CDF) → Not Recommended
Ibrutinib (relapsed/refractory Waldenström’s macroglobulinemia)
- First CDF entry (2017): Entered CDF with evidence from the single-arm PCYC-1118E trial (n=63) showing durable responses but immature survival data. Base-case ICER was £54,100/QALY –above NICE’s usual threshold – but high unmet need and patient testimony supported CDF inclusion.
- Reappraisal data (2022): Real-world outcomes from the SACT registry[15] (n=823 NHS patients) don’t include progression-free survival (PFS). The company therefore estimated PFS indirectly from time to treatment discontinuation, which NICE disagreed with. While updated trial data were available, these showed markedly better outcomes than the SACT cohort, likely due to differences in baseline characteristics (trial patients were younger and fitter; 24-month survival was 95% in trial 1118E vs 73% in SACT). In addition, comparative effectiveness vs chemo remained uncertain, and the updated ICERs still exceeded £30,000/QALY.
- Result: Recommendation withdrawn; ibrutinib was removed from the CDF and not commissioned for routine NHS use.
MAA for other drugs
Some non-cancer drugs with interim data are also reviewed by NICE. For that, we selected cases on the cutting edge of innovation:
- High-cost novel gene therapies
- Treatments for ultra-rare diseases affecting fewer than 1,000 patients
- First disease-modifying drug for Alzheimer’s
Narrowed scope may allow drugs to evolve from initial MAA
Zolgensma (SMA)
- First approval (2021, MAA): Initially in MAA due to small trials (START, STR1VE, SPR1NT) and uncertain long-term benefit
- Reappraisal (2023): New long-term data (LT-001, LT-002) showed sustained motor milestone gains for up to 4–7 years post-dose; many achieved independent sitting, standing, or walking
- Scope narrowing (2023, HST): Optimized to presymptomatic ≤12 months with ≤3 SMN2 copies, fitting HST’s ≤1,100 patient criteria and £300k/QALY threshold
- Result: Full routine commissioning—one-time therapy funded based on strong durability evidence, rare disease scope, and favorable ICER.
Zolgensma cleared NICE’s HST bar by narrowing its label to presymptomatic infants, which made the ultra-rare group (<1,100 in England) eligible for ICERs up to £300k/QALY.[16] NICE’s decision illustrates a willingness to fund expensive one-time therapies only when evidence is strong and scope is tight.
With strong scientific proof, even drugs approved via surrogate endpoint can bypass MAA.
Elafibranor (Primary Biliary Cholangitis)
- Approval (2024, STA): NICE judged the surrogate evidence to be robust enough to recommend routine commissioning. The committee highlighted that based on the scientific literature, serum levels of ALP and bilirubin are frequently used as primary efficacy endpoints in clinical trials for Primary Biliary Cholangitis (PBC). They are reliable surrogate markers of disease progression, powerful predictors of cholestatic injury, liver function, and transplant-free survival[17]
- Trial evidence: ELATIVE trial showed substantial ALP reductions and bilirubin normalization in a meaningful proportion of patients.
- Cost-effectiveness: £31,762/QALY vs UDCA; dominant vs OCA.
- Result: Routine commissioning–robust biomarker validation avoided need for managed access.
Surrogate endpoints are thought to add uncertainty to clinical outcomes, increasing the likelihood of rejection or placement in a MAA[18]. But for Elafibranor, strong scientific evidence allowed it to bypass managed access to win full approval.
Durability reigns value case
Hemgenix (Hemophilia B)
- Approval (2024, STA, MAA): One-time AAV gene therapy; list price £2.6M. MAA due to uncertainty in factor IX expression durability and clinical thresholds for restarting prophylaxis.
- Durability modelling: Cost-effective if benefit lasts >17 years; NHS benefit amplified by lifelong patient retention.
- Result: Managed access until 2030; value contingent on long-term real-world outcomes.
NICE raised concerns about the durability of factor IX expression and the clinical thresholds for restarting prophylaxis. This uncertainty has made NICE request data collection until 2030, when better value judgements can be made based on durability.
Incompatibility with healthcare infrastructure bars access through MAA
Leqembi (Alzheimer’s Disease patients with mild cognitive impairment)
- Rejection (2025): Rejected despite MHRA approval; NHS infrastructure unprepared for required diagnostics (PET-CT, CSF analysis, APOE4 genotyping) and MCI not formally recognized in NICE guidelines as a treatable stage.
- Clinical evidence:45-point improvement on CDR-SB vs placebo—below NICE’s minimal clinically important difference; limited data on progression from MCI to dementia.
- Cost: Company estimated £139/infusion; NHS England preferred £432 reflecting higher staffing and patient needs. This results in an ICER estimation of £93,769/QALY; even near-zero drug price scenarios failed to meet threshold when monitoring costs from infusions were included
- Result: Not recommended—insufficient infrastructure, modest benefit, and poor value for money.
Infrastructure gaps, low QALY benefit, and system-wide cost disputes led to NICE’s rejection of Leqembi. Products requiring an overhaul of a disease’s diagnostic pathway and significant healthcare system resources may result in ultimate rejection.
MAA summary
The UK’s managed access framework offers a pragmatic way to reimburse high-cost therapies while awaiting more evidence. Unlike a theoretical OBA, where installments could be tied to milestones (e.g., partial payment at years 3, 5, and beyond based on performance), an MAA is a one-time decision that commits the NHS to temporary funding. Budgets are buffered, as the Cancer Drugs Fund and Innovative Medicines Fund budgets are fixed. The examples above demonstrate that MAAs enable early patient access to promising treatments while establishing clear evidentiary requirements for broader NHS adoption. Depending on additional evidence, drugs may transition to full approval once clinical uncertainties are addressed, or be withdrawn when evidence fails to support their clinical benefit or cost-effectiveness. Ultimately, securing widespread patient access across the UK healthcare system requires biopharma companies to deliver robust evidence to justify broader adoption.
Translatability to the US
The UK’s experience with innovative pricing models offers important lessons for the U.S. Despite having seemingly ideal conditions, such as a centralized payer, national data systems, and established conditional reimbursement frameworks, the UK has migrated away from OBAs. Instead, the UK has adopted a more simplified MAA process. If OBAs haven’t flourished under the UK’s favorable structural conditions, they likely face even steeper challenges in the U.S., where there are major structural, regulatory, and infrastructure obstacles. (Exhibit 2)
Exhibit 2. Obstacles to an OBA in US
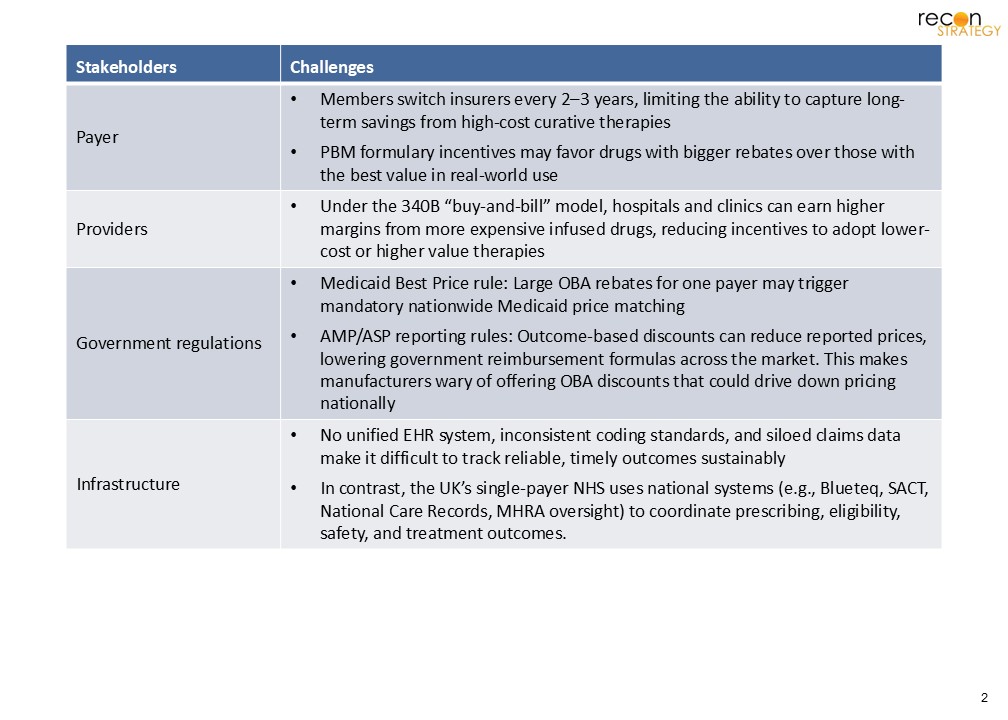
MFN policies complicate OBA adoption, as they currently have confidential pricing and rebates. In today’s environment, manufacturers may be less likely to enter into these arrangements where their lowest observable net price could be lowered, and subsequently used as a benchmark for other markets. However, there may still be room for a lighter touch, UK-style approach. Nonprofits like ICER (Institution for Clinical and Economic Review) could play larger roles by analyzing emerging data, flagging gaps, and making transparent assessments that feed into payer decisions. Rather than launching into more complex risk-sharing contracts, the first step could be building a consistent, credible process for acquiring real-world evidence, which can then trigger subsequent payer refinement for pricing and access.
Sources:
[1] DiPiro JT, Hoffman JM, Schweitzer P, et al. ASHP and ASHP Foundation Pharmacy Forecast 2024: Strategic Planning Guidance for Pharmacy Departments in Hospitals and Health Systems. Am J Health Syst Pharm. 2024;81(2):5-36. doi:10.1093/ajhp/zxad231
[2] https://www.spglobal.com/marketintelligence/en/mi/country-industry-forecasting.html?id=106598021
[3]https://www.nice.org.uk/guidance/ta215/documents/renal-cell-carcinoma-first-line-metastatic-pazopanib-final-appraisal-determination-guidance2
[4]NICE decisions are binding in England, generally adopted in Wales and Northern Ireland, but Scotland relies mainly on its own system https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-technology-appraisal-guidance
[5] https://www.nice.org.uk/news/blogs/should-nice-s-cost-effectiveness-thresholds-change-
[6] Earlier “end-of-life” criteria applied a £50,000 threshold
[7] https://www.valueinhealthjournal.com/article/S1098-3015(23)04936-7/fulltext#:~:text=Objectives,impact%20on%20committee%20decision%20making.
[8]https://www.ispor.org/docs/default-source/euro2023/hta301-how-eol-translates-to-severity-weighting-under-the-new-nice-methodologyispor23131602-pdf.pdf?sfvrsn=7fdeaed2_0
[9] To qualify, conditions must affect fewer than 1 in 50,000 people (~1,100) or no more than 300 patients in the licensed indication (≤500 across all indications), alongside high unmet need, severe impact, and potential for substantial clinical benefit
[10] Hale, George et al. “Flexibility in assessment of rare disease technologies via NICE’s single technology appraisal route: a thematic analysis.” Journal of comparative effectiveness research vol. 12,11 (2023): e230093. doi:10.57264/cer-2023-0093
[11] https://www.nice.org.uk/process/pmg37/chapter/highly-specialised-technologies
[12] https://www.england.nhs.uk/medicines-2/medicines-value-and-access/
[13]https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-technology-appraisal-guidance/data/cancer-appraisal-recommendations
[14] Median follow-up was 25.4 months; median progression-free survival (PFS) was 19.8 months.
[15] SACT tracks prescribing pattern and immediate treatment outcomes, such as regimen modification or discontinuation. They do not record specific interim outcomes, like progression free survivals.
[16] Pg 665, https://www.nice.org.uk/guidance/hst15/documents/committee-papers
[17] Pg 23, https://www.nice.org.uk/guidance/ta1016/documents/committee-papers
[18] Eg: Tecartus (Recommended, CDF); Tarpeyo (Optimized); Elrexfio (Optimized, CDF), ZTALMY (Not Recommended), Lunsumio (Not Recommended)
Appendix 1. Fate of oncology drugs post-CDF.
| Start | End | Number of cases | List of cases |
| Recommend (CDF) | Not Recommend | 2 | TA491 to TA795 TA519 to TA692 |
| Optimize | 10 | TA592 to TA802 TA529 to TA1021 TA756 to TA1018 TA483 to TA655 TA484 to TA713 TA416 to TA653 TA885 to TA939 TA600 to TA770 TA742 to TA1038 TA760 to TA1042 |
|
| Recommend | 7 | TA559 to TA872 TA588 to TA684 TA581 to TA780 TA490 to TA736 TA533 to TA766 TA554 to TA975 TA487 to TA796 |
|
| Optimized (CDF) | Optimized | 4 | TA446 to TA524 TA510 to TA783 TA505 to TA870 TA528 to TA784 |
| Recommend | 1 | TA540 to TA772 |