
Interest and investment in digital health has increased rapidly in recent years. Some digital health software is impactful enough that it requires FDA approval, but current regulatory pathways are slow and cumbersome for tech companies. In July 2017 the FDA announced a beta-test of a new pathway, the pre-certification program, which is intended to increase innovation and minimize barriers to market entry for digital health software. While early signs show the program will have these intended consequences, it also may create an uneven playing field for incumbent players relative to new, smaller organizations.
This is good news for established companies. Product managers at organizations that already have health tech on the market, that may have tried to avoid the FDA in the past, should be thinking about how they can push the capabilities of their products and how to best take advantage once the program is fully put in place. Smaller organizations should be thinking about starting now. Establish a presence, think about internal processes and KPIs so that they too can take advantage. Alternatively, smaller organizations should be thinking about how best to tackle areas of the market that larger companies may ignore, given their advantage.
It’s important to keep in mind that wellness apps and electronic medical records don’t require FDA clearance. This new program is designed to speed the market entry of “Software as a Medical Device” (SaMD). You can find the FDA definition of SaMD here.
One example of SaMD is digital therapeutics, which are software-based disease treatments that have gone through rigorous clinical trials and have been shown to have a substantial impact on the user’s health.
Within the digital therapeutics space, there is a range of products. One example of a company developing prescription digital therapeutics is Pear Therapeutics, which already has an FDA authorized product for non-Opioid Substance Use Disorder. They have products in development for other therapeutics areas including Schizophrenia and Parkinson’s Disease, all with the idea that the software would be paired with medical support from a doctor and sometimes other medicine.
Other examples of digital therapeutics include Omada Health’s behavior change program, or Natural Cycles’ app which is proven to prevent pregnancy. Digital therapeutics can also be particularly helpful for diagnosing conditions that may not be present constantly and can be hard to see in the doctor’s office, like the new Apple program that can detect an irregular heart rhythm.
Current FDA regulatory processes are complex and can delay a product’s market entry by months. Many companies have even moved early development out of the US to avoid FDA oversight. The pre-cert program allows the FDA to approve organizations based on their quality processes, instead of each software and software update. This allows software to get to consumers faster.
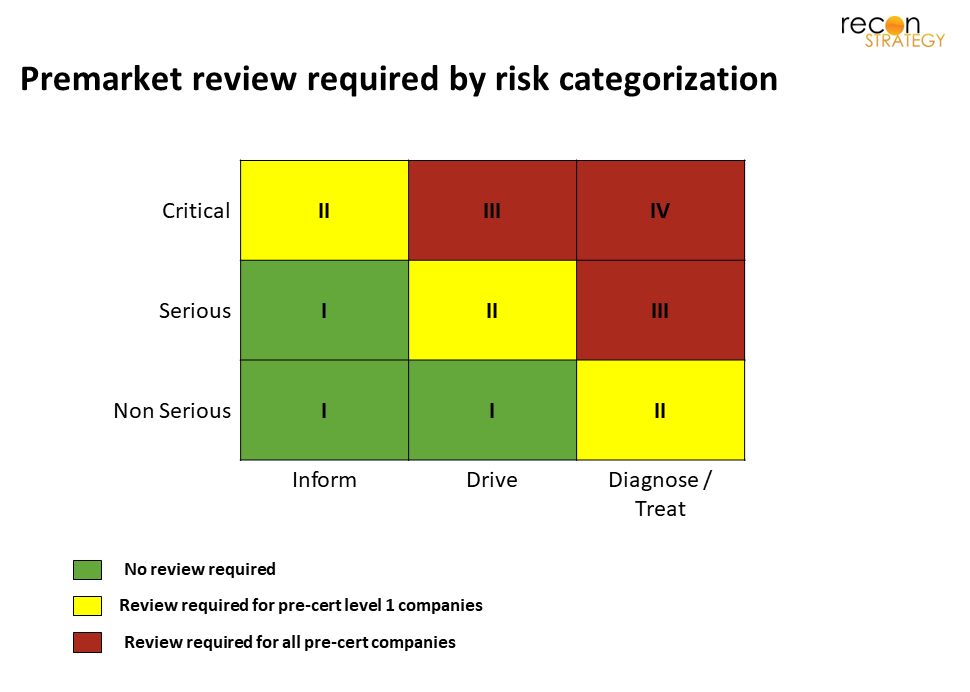
Some types of SaMD will still require review, depending on the risk type. More details are available in the model, but I’ve included a summary table of risk types below.

In order to participate in the pre-cert program, companies must show competence in the 5 “excellence principles”, which are: Product Quality, Patient Safety, Clinical Responsibility, Cybersecurity Responsibility and Proactive Culture. However, the FDA has created two levels of certification, Level 1 is for companies of any size and history, and Level 2 is for companies with a proven track record for developing SaMD.
The required level of review depends not only on the risk type, but also on the company’s level of certification. The details can be seen here:
Historically, the burdensome FDA clearance process has pushed companies to limit the capabilities of their software to avoid the need for regulation. If done properly, this new process should allow for developers to move away from wellness apps, and towards impactful digital therapeutics. There are already some signs of this – FitBit has historically shied away from seeking FDA clearance for its wearables but is now one of the partner companies developing the pre-cert program model. Tidepool has written extensively on their blog about their positive experience working with the FDA to develop and implement the new model.
Additionally, the pre-cert program may enhance the relationship between software companies and consumers. Since software can be released without FDA review, the importance of post-market feedback increases. Pre-certified organizations are expected to be responsive to consumer feedback. This meshes well with the overall trend in healthcare towards increased consumer choice and engagement.
In order to maintain some control, the FDA has recognized that some vetting still needs to be done for software in higher risk categories. It has also recognized the value of a proven track record for developing in the space.
The unintended consequence of these limitations is that companies with no track record have a relative barrier to entry in certain risk categories. Much like the days pre-pre-cert, Level 1 companies may choose to limit which risk categories they take on. This allows Level 2 companies to have an advantage in three domains. Without requiring review, they can:
- Develop software to inform clinical management for critical diseases (not just serious)
- Develop software to drive clinical management for serious diseases
- Develop technologies to diagnose or treat patients with non-serious diseases.
In the chart below, the arrows depict areas where if a technology is on the margin, a Level 2 company has a relative advantage to move from green to yellow, while a Level 1 company may choose to stay in the green area where no review is required.

While the FDA has emphasized their desire to spur innovation and to encourage companies of all sizes to participate in the pre-cert program, it seems that larger companies could still maintain an advantage.
The key will be for smaller companies, or companies with limited exposure to the healthcare space, to establish their track record early. Also, developers who have gone through the process recommend communicating with the FDA early and frequently. Another option is to focus in the “Type I” space, which larger companies may ignore. The FDA has also been encouraging public comment on the model, so interested parties should feel they have a chance to impact the future process.